Il campo d'indagine della genetica di popolazioni è molto vasto e
interdisciplinare e si intreccia con con quello di numerose altre
scienze: la biologia molecolare, la genetica, l'ecologia, la biologia
evolutiva, la sistematica, la storia naturale, il miglioramento
genetico, la conservazione delle specie e degli ambienti naturali, la
genetica umana, la sociologia, la matematica e la statistica.
Molti ricercatori sono concordi nel dire che questi anni sono davvero straordinari per la genetica di popolazione, poichè la genetica molecolare sta contribuendo ad uno sviluppo davvero esponenziale e la genetica di popolazione finalmente ha a disposizione strumenti e dati mai avuti prima.
Semplificando un po' si può dire che la genetica di
popolazioni studia le modalità secondo cui le leggi di Mendel e
gli altri principi della genetica si applicano alle popolazioni. Tale
approccio è essenziale per una corretta comprensione
dell'evoluzione.
L'evoluzione, a livello di base, è definita come i cambiamenti progressivi che subiscono le frequenze alleliche nelle popolazioni.
Spiegare e modellizzare l'evoluzione, anche se definita in un modo semplice come sopra, può sembrare un compito tutto sommato facile. In realtà i fattori molecolari, genetici ed ecologici che entrano in campo sono tali e tanti che rendono il compito molto difficile.
Uno dei principi della genetica di popolazioni è che non ci
può essere evoluzione se non c'è variazione genetica. In
sostanza l'evoluzione deve avere ``materia prima'' su cui operare.
Compiti della genetica di popolazioni:
Già sul primo di questi compiti c'è stata (e tuttora c'è) molta attitivà di ricerca e non siamo tutt'oggi in grado di dare una risposta precisa. Sugli ultimi due punti poi c'è stato un dibattito molto acceso e mai sopito tra i biologi evoluzionistici. Esamineremo in maggior dettaglio tutti questi aspetti, partendo dal primo.
Esistono numerosi metodi per analizzare la variabilità genetica. Alcuni metodi hanno avuto più successo di altri; alcuni sono stati importanti in passato, altri saranno importanti in futuro; alcuni metodi sono facili, altri sono più difficili e lunghi. Noi faremo una rassegna di alcuni di questi metodi, cercando di capirne il funzionamento, i pregi e i difetti.
La variabilità genetica delle popolazioni naturali è
misurata mediante l'uso di marcatori genetici.
Prima di iniziare la rassegna però diamo la definizione di
marcatore genetico e cerchiamo di chiarire quali siano le
caratteristiche di un marcatore genetico ideale
Caratteristiche ideali di un marcatore genetico:
In realtà non esistono ancora marcatori genetici che hanno tutte queste caratteristiche. La scelta del tipo di marcatore da utilizzare dipenderà dagli scopi della ricerca e dai mezzi a disposizione.
Rispetto al genoma studiato distinguiamo in:
questi marcatori differiscono anche per il tipo di eredità (paterna e/o materna).
Rispetto al metodo impiegato distinguiamo in:
È uno dei metodi tuttora più usati, anche se in modo sempre minore. Nasce alla fine degli anni '60 ed ha prodotto un'enorme quantità di dati. È stato un metodo rivoluzionario perchè ha permesso, per la prima volta, la verifica delle ipotesi e dei modelli della teoria evoluzionistica.

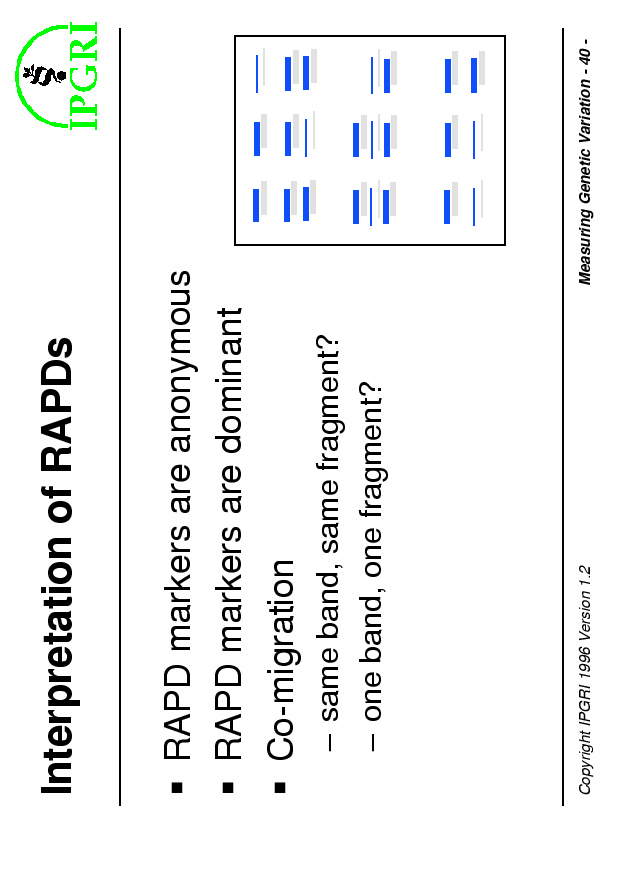

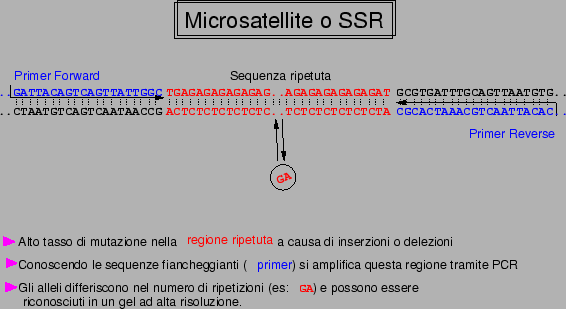
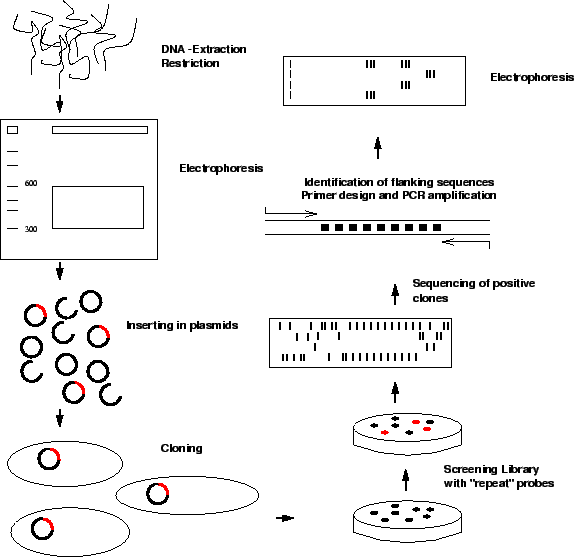
Pro
Contro
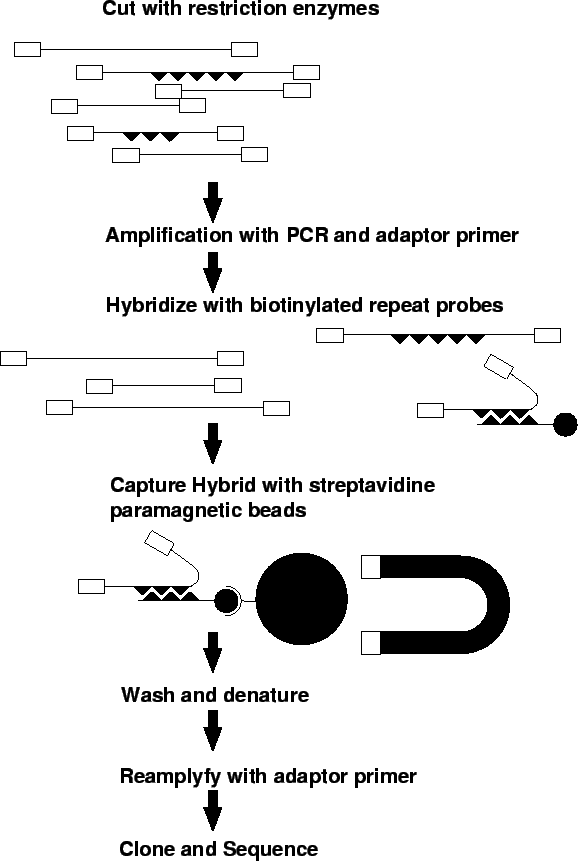
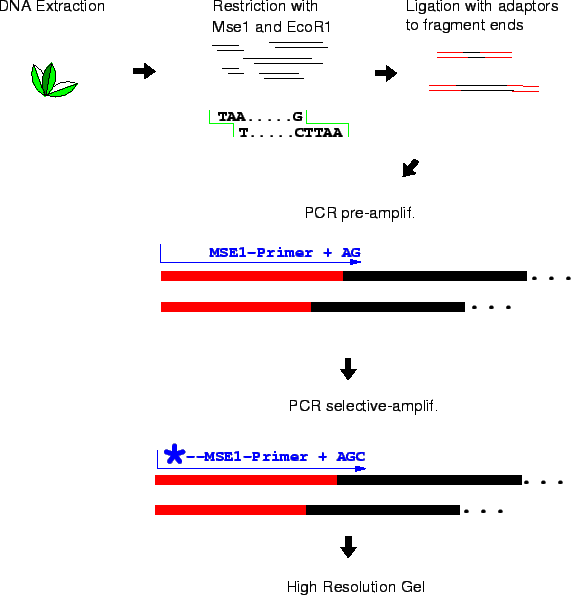
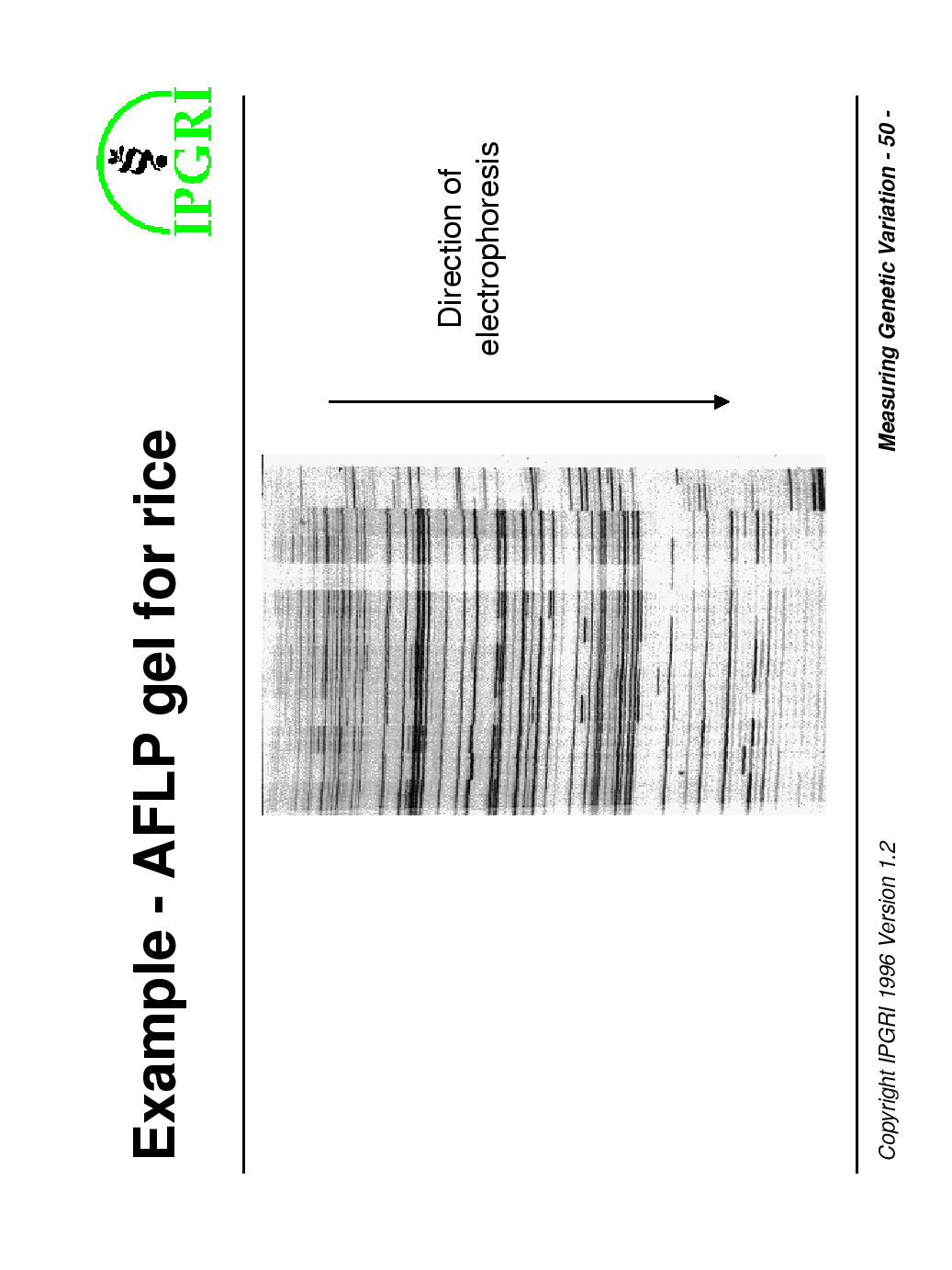
La tecnica AFLP (Amplified Fragment Length Polymorphism) è molto sensibile nel trovare polimorfismi in tutto il genoma e sta diventando sempre più popolare. La procedura è stata pubblicata per la prima volta nel 1995. Le fasi sono le seguenti:
La scelta degli enzimi di restrizione e dei due-tre nucleotidi arbitrari in terminazione 3' permette una scelta elevatissima di combinazioni che amplificheranno tratti diversi di DNA. Normalmente con una combinazione enzima-estensione si riescono ad amplificare a 10-20 (talvolta 100) frammenti (potenzialmente loci) diversi.
Pro
Contro
Il principio di Hardy-Weinberg è uno dei principi più importanti per la genetica di popolazioni. È una principio molto semplice e intuitivo che fornisce una sorta di ``modello nullo'' contro cui verificare i dati riscontrati nel mondo reale.
Il principio di Hardy-Weinberg permette di stimare, se tutte le assunzioni sono vere, le frequenze genotipiche partendo dalle frequenze alleliche.
Una deviazione significativa dalle previsioni dell'equilibrio di Hardy-Weinberg ci dice che almeno una delle assunzioni non è vera. Sta allo sperimentatore cercare di capirne il perchè.
Ma vediamo cosa prevede il principio di Hardy-Weinberg nel caso di una popolazione con due alleli (A e a):
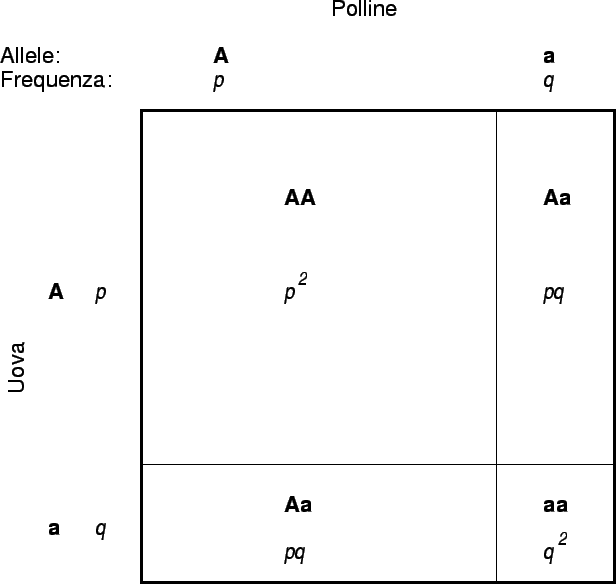
Il principio di Hardy-Weinberg stabilisce quindi che se gli alleli (A e a) hanno rispettivamente frequenza p e q, le frequenze genotipiche attese saranno:
Se tutte le assunzioni sono verificate l'equilibrio di Hardy-Weinberg si raggiunge in una sola generazione.
È possibile dimostrare che il principio di Hardy-Weinberg è vero anche se partiamo dai genotipi e dalle loro frequenze invece che dai gameti come nella figura precedente.
Domande:

Nella lezioni precedenti abbiamo imparato a calcolare le frequenze alleliche e le frequenze genotipiche. Abbiamo anche imparato a calcolare le frequenze genotipiche attese in base all'equilibrio di Hardy-Weinberg.
Se si analizzano più loci, l'eterozigosi osservata media è la media delle eterozigosi osservate per ciascun locus.
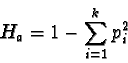 |
(5.1) |
Anche in questo caso, se si analizzano più loci, l'eterozigosi attesa media è la media delle eterozigosi attese per ciascuno dei vari loci.
L'eterozigosi attesa è una misura molto usata per valutare la variabilità genetica di una popolazione. Se estraggo casualmente due alleli dalla popolazione, misura la probabilità di che i due alleli siano differenti.
È massima quando le frequenze degli alleli sono bilanciate (es: 0.5 e 0.5 nel caso di due alleli, 0.33, 0.33 e 0.33 nel caso di tre alleli, ecc) e in generale aumenta all'aumentare del numero di alleli.
Eventuali deviazioni dall'equilibrio di Hardy-Weinberg vengono spesso definite semplicemente come un eccesso o un difetto di omozigoti (o di eterozigoti) rispetto all'atteso.
In questi casi si fa ricorso ad un solo indice numerico detto indice di fissazione (F). Nel caso di due alleli possiamo capire da dove deriva la definizione di F:
| Genotipo | Incrocio | Inincrocio |
|---|---|---|
| casuale | (o Esoincrocio) | |
| AA | p2 | p2 + pqF |
| Aa | 2pq | 2pq - 2pqF |
| aa | q2 | q2 + pqF |
Concentriamoci solo sulla riga degli eterozigoti Aa,
sostituendo il termine 2pq con il più generale Ha e
ammettendo che il termine
2pq - 2pqF corrisponda all'eterozigosi
osservata (Ho), possiamo definire F come:
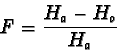
cioè F assume valori positivi in caso di eccesso di omozigoti, mentre assume valori negativi in caso di difetto di omozigoti.
Nel caso di eccesso di omozigoti (come nel caso dell'inincrocio), Fpuò essere inteso come quella frazione di eterozigoti ``trasformati'' in omozigoti dall'incrocio non casuale.
Vediamo ora il comportamento di alcune popolazioni virtuali tramite il programma ape. Il programma ape legge un file di configurazione (es: ape.conf) di cui riportiamo qui un esempio:
LatoBosco = 20 LatoNeighbourhood = 5 NGenerations = 100 EveryNGenerations = 10 NLoci = 4 Locus1 = 0.50 Locus2 = 0.40 Locus3 = 0.60 Locus3 = 0.90
Il seguente diagramma spiega come avviene la riproduzione da una generazione all'altra.
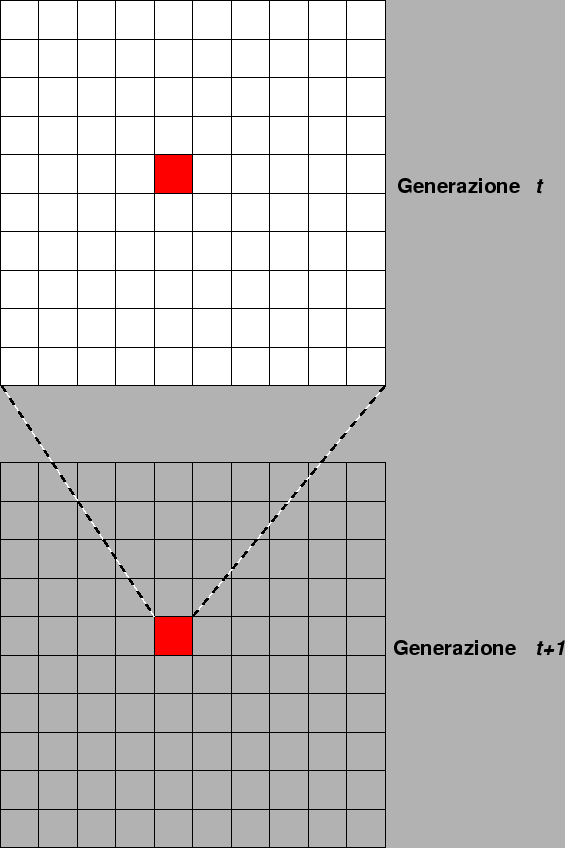
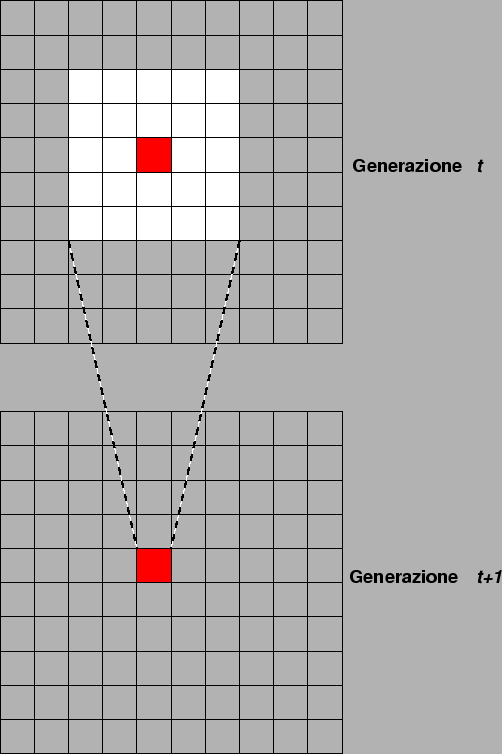
Fate correre il programma ape per un numero sufficiente di generazioni, cambiando le dimensioni delle popolazioni, quindi compilate la seguente tabella. In ogni riquadro potete inserire le seguenti caratteristiche che descrivono l'andamento del parametro nel tempo:
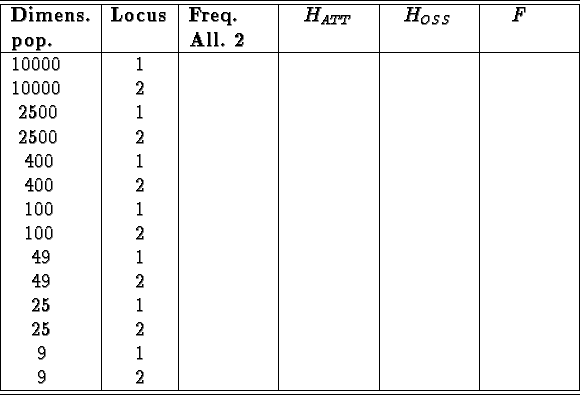
Quindi rifate la simulazione con N=10000 imponendo un
LatoNeighbourhood = 3 cioè restringendo la dispersione
pollinica all'interno della popolazione.
Gli effetti sulle frequenze alleliche che abbiamo osservato al diminuire della dimensione della popolazione sono tipiche del fenomeno chiamato deriva genetica. Perchè la deriva genetica abbia effetto occorre che le popolazioni siano isolate e evolvano indipendentemente una dall'altra. Gli effetti della deriva genetica sono tanto più marcati e visibili quanto più piccola è la popolazione. Notare che in nessuna parte della simulazione un allele viene favorito rispetto ad un altro (la selezione non agisce!).
Riassumiamo gli effetti della deriva genetica
| (5.2) |
Ora cambiamo scala spaziale alla quale osserviamo gli effetti della deriva genetica: consideriamo il caso in cui l'habitat subisca una frammentazione (evento purtroppo molto comune) e quindi immaginiamo che la nostra popolazione venga a sua volta frammentata in due sotto-popolazioni. Ciascuna sotto-popolazione è, almeno in prima assunzione, isolata dalle altre ed è sottoposta a deriva genetica in maniera tanto più marcata quanto più piccola è la sotto-popolazione.
Immaginiamo che le sotto-popolazioni siano due, ciascuna di 12 individui, che esistano solo due alleli, e che l'incrocio sia casuale all'interno di ciascuna popolzione. La seguente figura esemplifica l'andamento atteso nel tempo delle due popolazioni:
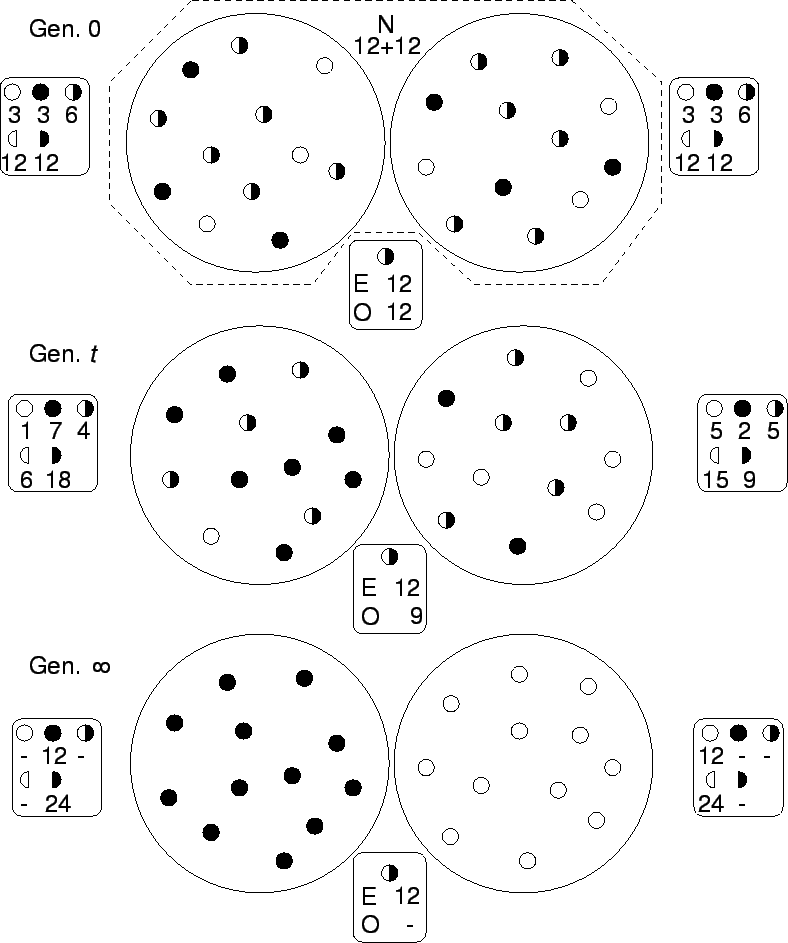
Man mano che il tempo progredisce le due popolazioni si differenziano sempre di più (considerando che nella realtà ci sono molte sotto-popolazioni, molti loci implicati e molti alleli, è rarissimo che le due popolazioni evolvano casualmente allo stesso modo). Man mano che le sotto-popolazioni si differenziano, anche se al suo interno ciascuna sotto-popolazione può essere in equilibrio di Hardy-Weinberg, nella popolazione totale si avrà un progressivo eccesso di omozigoti.
In genetica di popolazioni si usa molto spesso un paramentro, l'FST, che misura appunto l'eccesso di omozigoti dovuto alla frammentazione della popolazione totale in tante piccole sotto-popolazioni. L'FST è quindi un indice di ``distanza genetica'' fra le popolazioni (più le popolazioni sono differenziate, maggiore sarà l'eccesso di omozigoti dovuto alla frammentazione, maggiore sarà l'FST).
L'eventuale eccesso di omozigoti all'interno delle
popolazioni, causato per esempio da inincrocio, sarà
misurato da un indice diverso: l'FIS.
Entrambi rendono conto del generale eccesso di omozigoti nella popolazione totale che sarà misurato dall'FIT.
Avremo, per ogni locus analizzato, un'eterozigosi osservata e attesa in ciasuna delle sottopopolazioni analizzate e avremo eterozigosi media (osservata ed attesa) per la popolazione totale.
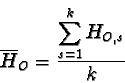 |
(5.3) |
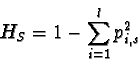 |
(5.4) |
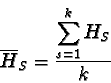 |
(5.5) |
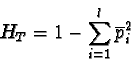 |
(5.6) |
dove
![]() è la frequenza media dell'allele i (pesata
per la dimensione del campione) in tutte le popolazioni (oppure può
essere inteso come la frequenza dell'allele i nella popolazione
totale ottenuta sommando tutti i campioni di tutte le
sottopopolazioni)
è la frequenza media dell'allele i (pesata
per la dimensione del campione) in tutte le popolazioni (oppure può
essere inteso come la frequenza dell'allele i nella popolazione
totale ottenuta sommando tutti i campioni di tutte le
sottopopolazioni)
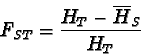 |
(5.7) |
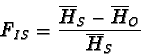 |
(5.8) |
Il flusso genico tra popolazioni (migrazioni di geni via polline o seme da una popolazione ad un'altra) è una forza che tende a rimescolare i geni fra le sotto-popolazioni, quindi tende a uniformare, rendere più simili le popolazioni.
Il flusso genico può essere quindi inteso come la forza contraria alla deriva genetica. Cioè mentre la deriva genetica tende a diversificare le popolazioni, il flusso genico tende a renderle più simili.
Quindi l'FST può essere usato, assumendo che la storia delle sotto-popolazioni sia simile e che la selezione non operi, come una misura del flusso genico avvenuto fra le popolazioni. Un maggior flusso genico implica una minore differenziazione delle popolazioni e quindi un più basso FST.
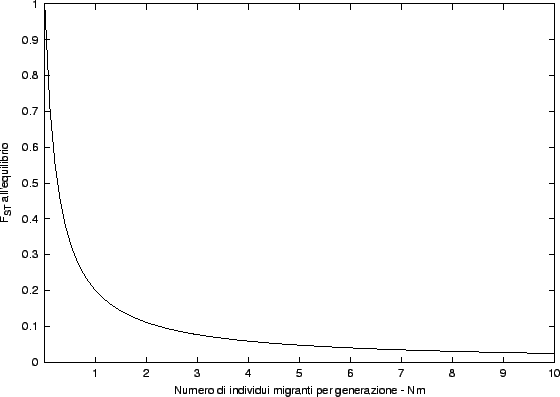
Una dei modelli più usati in genetica di popolazioni è il modello dell'isola di Wright in cui si ipotizza che una popolazione su un'isola subisca l'immigrazione da un'ipotetica popolazione molto più grande situata sul continente. In questo caso il modello porta alla seguente relazione:
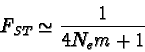 |
(5.9) |
in cui Ne è la dimensione effettiva della popolazione, m è la percentuale di individui migranti per generazione, quindi Nem sono il numero di individui migranti per generazione. Dal seguente grafico, molti hanno dedotto che bastano pochissimi individui migranti per generazione per impedire che le popolazioni si differenzino in modo significativo.
Se una popolazione si differenzia in modo significativo dalle altre e rimane in isolamento per lungo tempo, possono instaurarsi delle barriere riproduttive che impediscono il flusso genico cioè la riproduzione (o la produzione di prole feconda) fra la popolazione isolata e le altre popolazioni. In questo caso, anche se la popolazione tornasse ad essere non-isolata, il flusso genico sarebbe assente; si sarebbe così generata una nuova specie.
Una delle applicazioni più riscontrate nella letteratura scientifica di genetica di popolazioni di piante e animali è il confronto di popolazioni (sotto-popolazioni) diverse. Si possono confrontare diversi parametri che descrivono le popolazioni ma sicuramente il paramentro più usato sono le frequenze alleliche.
Il dato di partenza è una tabella molto simile alla seguente dove per ogni locus e per ogni allele sono riportate le frequenze alleliche. Queste frequenze sono tanto più attendibili quanto più la dimensione del campione è grande.
Nella seguente tabella (vera ma con qualche dato appositamente cambiato) immaginiamo di avere campionato le popolazioni in quattro regioni differenti per diversi marcatori allozimici.
STAZ REGIONE ADH.1 ADH.2 ADH.3 DIA.1 DIA.2 DIA.3 GDH.1 GDH.2 GDH.3
CGA Reg.1 0.0435 0.7609 0.1956 0.0000 1.0000 0.0000 0.0000 0.3636 0.6364
CRO Reg.1 0.0000 0.6395 0.3605 0.0000 0.8659 0.1341 0.0000 0.2875 0.7125
FSE Reg.2 0.0299 0.9478 0.0224 0.0484 0.9355 0.0161 0.1102 0.7966 0.0932
FST Reg.2 0.0000 0.8583 0.1417 0.0656 0.9344 0.0000 0.0417 0.8417 0.1167
FTR Reg.2 0.0000 0.8958 0.1042 0.1985 0.8015 0.0000 0.0000 0.7705 0.2295
FLM Reg.2 0.0643 0.7786 0.1571 0.1104 0.8831 0.0065 0.0682 0.7364 0.1954
FRO Reg.2 0.0250 0.9500 0.0250 0.1328 0.8672 0.0000 0.0909 0.7727 0.1364
RLM Reg.3 0.1364 0.8636 0.0000 0.3500 0.6500 0.0000 0.1286 0.5500 0.3214
RRO Reg.3 0.2000 0.7571 0.0429 0.1328 0.8672 0.0000 0.0078 0.7188 0.2734
RCA Reg.3 0.0763 0.8220 0.1017 0.3968 0.5317 0.0714 0.1667 0.7778 0.0556
RMA Reg.3 0.0530 0.8561 0.0909 0.6129 0.3871 0.0000 0.2119 0.6356 0.1525
RSA Reg.3 0.2027 0.6081 0.1892 0.7014 0.2014 0.0972 0.0250 0.9000 0.0750
LCH Reg.4 0.1250 0.8333 0.0417 0.0741 0.6574 0.2685 0.1442 0.6154 0.2404
LVB Reg.4 0.1042 0.8854 0.0104 0.0667 0.7667 0.1667 0.0000 1.0000 0.0000
STAZ REGIONE GOT.1 GOT.2 GOT.3 G3PDH.1 G3PDH.2 IDH.1 IDH.2 IDH.3 IDH.4
CGA Reg.1 0.1071 0.7500 0.1429 1.0000 0.0000 0.0000 0.1512 0.8488 0.0000
CRO Reg.1 0.3140 0.6861 0.0000 1.0000 0.0000 0.1463 0.1341 0.7195 0.0000
FSE Reg.2 0.0379 0.9621 0.0000 1.0000 0.0000 0.0000 0.3095 0.6270 0.0635
FST Reg.2 0.0820 0.8197 0.0984 1.0000 0.0000 0.0000 0.2708 0.6806 0.0486
FTR Reg.2 0.0000 0.8191 0.1809 1.0000 0.0000 0.0000 0.2344 0.7656 0.0000
FLM Reg.2 0.0454 0.9318 0.0227 1.0000 0.0000 0.0066 0.1974 0.6526 0.1434
FRO Reg.2 0.1742 0.7576 0.0682 1.0000 0.0000 0.0000 0.2295 0.7377 0.0328
RLM Reg.3 0.1119 0.8508 0.0373 1.0000 0.0000 0.1154 0.2692 0.6154 0.0000
RRO Reg.3 0.1136 0.8864 0.0000 1.0000 0.0000 0.0000 0.0968 0.9032 0.0000
RCA Reg.3 0.2857 0.7143 0.0000 1.0000 0.0000 0.0323 0.1452 0.8145 0.0081
RMA Reg.3 0.1250 0.7734 0.1016 1.0000 0.0000 0.0000 0.0583 0.7583 0.1833
RSA Reg.3 0.1441 0.8559 0.0000 1.0000 0.0000 0.0000 0.2353 0.6471 0.1177
LCH Reg.4 0.0755 0.9245 0.0000 0.8737 0.1263 0.0000 0.0100 0.9600 0.0300
LVB Reg.4 0.0000 1.0000 0.0000 0.8667 0.1333 0.0000 0.0000 1.0000 0.0000
STAZ REGIONE MDH.1 MDH.2 MDH.3 SPGDH.1 SPGDH.2 SPGDH.3 SPGDH.4 PGI.1 PGI.2
CGA Reg.1 0.1122 0.8878 0.0000 0.0652 0.0217 0.9130 0.0000 0.0000 0.0000
CRO Reg.1 0.3095 0.4881 0.2024 0.0732 0.2927 0.6341 0.0000 0.0000 0.0000
FSE Reg.2 0.0484 0.7984 0.1532 0.0000 0.1967 0.8033 0.0000 0.0076 0.8864
FST Reg.2 0.0608 0.9324 0.0068 0.0000 0.0423 0.8873 0.0704 0.0205 0.8836
FTR Reg.2 0.0968 0.8790 0.0242 0.0000 0.2339 0.7661 0.0000 0.0746 0.8955
FLM Reg.2 0.0125 0.9812 0.0063 0.0000 0.1467 0.8533 0.0000 0.0688 0.9250
FRO Reg.2 0.0606 0.9318 0.0076 0.0073 0.1588 0.7456 0.0882 0.0333 0.9417
RLM Reg.3 0.1500 0.7071 0.1429 0.0000 0.1111 0.7936 0.0952 0.0328 0.9426
RRO Reg.3 0.1525 0.8220 0.0254 0.0000 0.0000 0.9701 0.0299 0.1111 0.8413
RCA Reg.3 0.1508 0.8254 0.0238 0.0000 0.1557 0.8443 0.0000 0.0593 0.8644
RMA Reg.3 0.2866 0.7060 0.0075 0.0000 0.3306 0.6452 0.0242 0.0313 0.9297
RSA Reg.3 0.1014 0.8551 0.0435 0.0000 0.2174 0.7681 0.0145 0.1030 0.8091
LCH Reg.4 0.0481 0.7308 0.2212 0.0096 0.0289 0.9231 0.0385 0.0536 0.9018
LVB Reg.4 0.1429 0.8469 0.0102 0.0000 0.0488 0.9512 0.0000 0.0227 0.9546
STAZ REGIONE PGI.3 PGI.4 PGM.1 PGM.2 PGM.3 SKDH.1 SKDH.2 SKDH.3
CGA Reg.1 0.1531 0.8469 0.0400 0.4200 0.5400 0.0000 0.8125 0.1875
CRO Reg.1 0.3293 0.6707 0.0000 0.6098 0.3902 0.2250 0.6625 0.1125
FSE Reg.2 0.1061 0.0000 0.1087 0.8406 0.0507 0.0000 0.9710 0.0290
FST Reg.2 0.0959 0.0000 0.1894 0.7500 0.0606 0.0156 0.9375 0.0469
FTR Reg.2 0.0299 0.0000 0.1849 0.7808 0.0343 0.0678 0.8729 0.0593
FLM Reg.2 0.0063 0.0000 0.1400 0.7600 0.1000 0.0068 0.9257 0.0676
FRO Reg.2 0.0250 0.0000 0.0809 0.8456 0.0735 0.0221 0.8971 0.0809
RLM Reg.3 0.0246 0.0000 0.1045 0.7985 0.0970 0.0333 0.9167 0.0500
RRO Reg.3 0.0476 0.0000 0.0753 0.8836 0.0411 0.1652 0.7913 0.0435
RCA Reg.3 0.0763 0.0000 0.0577 0.9039 0.0385 0.0476 0.9127 0.0397
RMA Reg.3 0.0391 0.0000 0.0833 0.8409 0.0758 0.0423 0.8451 0.1127
RSA Reg.3 0.0879 0.0000 0.1884 0.7536 0.0580 0.0469 0.7734 0.1797
LCH Reg.4 0.0446 0.0000 0.0200 0.8800 0.1000 0.0000 0.9727 0.0273
LVB Reg.4 0.0227 0.0000 0.2391 0.7609 0.0000 0.0000 0.7000 0.3000
Per svolgere questo compito sono state inventate diverse misure di distanza genetica che misurano la differenza genetica fra le popolazioni confrontando le frequenze alleliche tenendo conto di tutti i loci analizzati.
Una misura di distanza che può essere usata in questo caso è ovviamente l'FST calcolato per ogni coppia possibile di popolazioni, ma più spesso vengono usate altre distanze.
Una delle distanze genetiche che ha avuto maggior successo è la
distanza genetica di Nei:
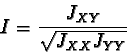 |
(5.10) |
Di solito il lavoro consiste nel calcolare una matrice di distanze genetiche in cui ogni popolazione viene confrontata con tutte le altre.
| Pop. A | Pop. B | Pop. C | Pop. D | Pop. E | Pop. ... | |
|---|---|---|---|---|---|---|
| Pop. A | 0 | 0.075 | 0.077 | 0.051 | 0.152 | ... |
| Pop. B | 0 | 0.080 | 0.048 | 0.142 | ... | |
| Pop. C | 0 | 0.045 | 0.176 | ... | ||
| Pop. D | 0 | 0.186 | ... | |||
| Pop. E | 0 | ... | ||||
| Pop. ... |
Da una matrice di distanze genetiche si procede poi a costruire un albero filogenetico che ricostruisce e rappresenta in modo grafico le differenze genetiche misurate.
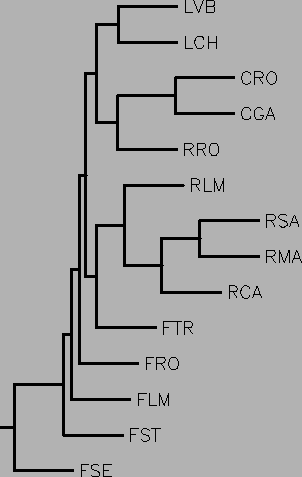
Esistono numerosi metodi per stimare le distanze genetiche e metodi per costruire gli alberi. Un metodo può produrre risultati leggermente o sostanzialmente diversi dagli altri. Ogni metodo ha pregi e difetti e soprattutto è stato sviluppato pensando ad un modello di evoluzione delle popolazioni. In questa sede non ci dilunghiamo su questi metodi ma riteniamo giusto concludere che la scelta del modello non dovrebbe essere fatta in base alla ``bellezza'' del risultato, ma dovrebbe essere fatta, prima di vederne il risultato, in base al modello di evoluzione che pensiamo abbia agito sulle nostre popolazioni.
Altri metodi, come l'analisi delle componenti principali
riassumono i dati delle frequenze alleliche in due o tre variabili
``principali'' che, appunto, sintetizzano i dati. Le nuove variabili
sintetiche sono calcolate sempre partendo dalle frequenze alleliche,
e tramite algoritmi dell'algebra lineare si calcolano delle nuove
variabili che sono una combinazione lineare delle frequenze
alleliche originali (pi)
| (5.11) |
Le nuove variabili sintetiche sono quindi una ``somma'' delle frequenze originali, ma ciascuna frequenza avrà un peso che dipende dal coefficiente ai, che può avere valori tra -1 e +1.
Una volta trovati valori i dei coefficienti ai si possono sostituire per ogni stazione le frequenze alleliche nella formula precedente e si ricava il valore di Y1 per ogni popolazione. Si può fare lo stesso per una seconda variabile sintetica Y2, che avrà i coefficienti ai diversi dalla prima, e quindi fare un grafico con le due Y come ordinata e come ascissa.
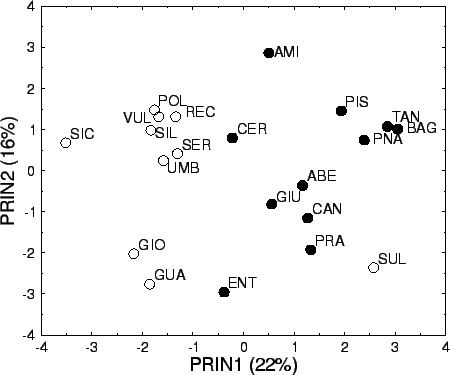
Il risultato che si presenta piuttosto di frequente è che il grafico delle componenti principali o l'albero in qualche modo ``ripercorrono'' la geografia delle popolazioni campionate. Cioè popolazioni vicine geograficamente andranno a finire vicine nel grafico o nell'albero. In sostanza la distanza genetica dipende dalla distanza geografica, un risultato spesso attribuito alle vicende storiche delle popolazioni esaminate.
Con selezione naturale si intende l'insieme dei fattori che tendono a favorire o sfavorire un dato genotipo e quindi ad aumentare o diminuire le frequenze degli alleli che lo compongono.
Una delle assunzioni dell'equilibrio di Hardy-Weinberg era l'assenza di selezione e si ammetteva quindi che tutti i genotipi avessero una fitness uguale a 1. Inoltre si ammetteva che la popolazione avesse una dimensione tendente all'infinito e non variasse nel tempo.
Si definisce la fitness come fitness relativa ad una fitness media della popolazione.
Se la fitness di tutti i genotipi è uguale allora non ci sono cambiamenti nelle frequenze alleliche. Ma se uno dei genotipi è sfavorito allora la sua fitnessa sarà minore. Nel caso di un allele recessivo letale avremo che:
| Genotipo | Frequenza | Fitness |
|---|---|---|
| AA | p2 | 1 |
| Aa | 2pq | 1 |
| aa | q2 | 0 |
Alla generazione successiva la frequenza dell'allele a sarà
data dal prodotto della frequenza del genotipo omozigote ![]() la
sua fitness + 1/2 della frequenza del genotipo eterozigote
la
sua fitness + 1/2 della frequenza del genotipo eterozigote ![]() la sua fitness, il tutto diviso per la fitness media:
la sua fitness, il tutto diviso per la fitness media:
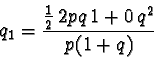
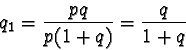
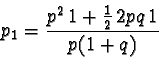
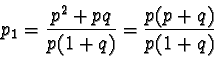
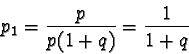
Nel caso generale si può scrivere la seguente tabella:
| Genotipo | Frequenza | Fitness |
|---|---|---|
| AA | p2 | w11 |
| Aa | 2pq | w12 |
| aa | q2 | w22 |
dove con w indichiamo la fitness relativa, che sarà maggiore di 1 (w = 1 + s) nel caso in cui il genotipo sia favorito dalla selezione e l'indice di selezione s sarà positivo, mentre wsarà minore di 1 nel caso in cui il genotipo sia sfavorito (snegativo).
La formula che esprime la frequenza (p') di un allele in una data
generazione in funzione della frequenza alla generazione precedente
(p) sarà data da:
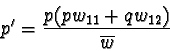
dove
![]() è la fitness media ed è data semplicemente da:
è la fitness media ed è data semplicemente da:
![\begin{displaymath}\Delta p = \frac{pq[p(w_{11} - w_{12}) + q (w_{12} -
w_{22})]}{\overline{w}}\end{displaymath}](img84.gif)
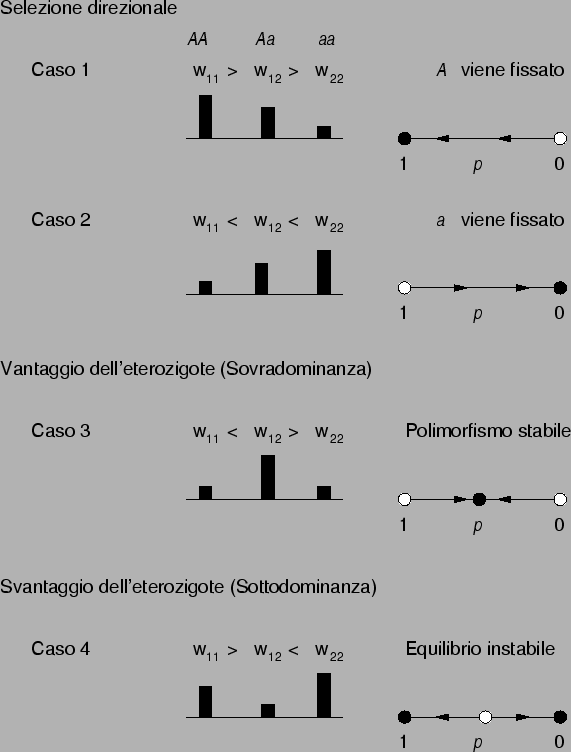
Sono teoricamente possibili diversi punti di equilibrio che si
verificano quando
![]() , cioè le frequenze non si
modificano nel tempo. I punti di equilibrio dipenderanno dalle fitness
dei vari genotipi. Alcuni equilibri sono banali e si verificano quando
p o q sono uguali a 0 o 1. Esistono altri due punti di equilibrio,
uno stabile quando c'è il vantaggio dell'eterozigote e uno
non stabile quando l'eterozigote è svantaggiato.
, cioè le frequenze non si
modificano nel tempo. I punti di equilibrio dipenderanno dalle fitness
dei vari genotipi. Alcuni equilibri sono banali e si verificano quando
p o q sono uguali a 0 o 1. Esistono altri due punti di equilibrio,
uno stabile quando c'è il vantaggio dell'eterozigote e uno
non stabile quando l'eterozigote è svantaggiato.
| Locus | Genotipo | Pop. A | Pop. B |
|---|---|---|---|
| Locus 1 | 11 | 6 | 3 |
| 12 | 8 | 8 | |
| 22 | 6 | 9 | |
| Locus 2 | 11 | 2 | 1 |
| 12 | 5 | 4 | |
| 13 | 2 | 3 | |
| 22 | 4 | 2 | |
| 23 | 4 | 4 | |
| 33 | 3 | 6 |
Come abbiamo visto il flusso genico è una delle forze evolutive più importanti nel determinare la struttura genetica delle popolazioni. Gli effetti del flusso genico dipenderanno da:
Diamo alcune definizioni utili nel definire le modalità riproduttive:
Le modalità di studio del flusso genico sono diverse. Nelle piante possiamo avere flusso genico attraverso:
Solo la seconda e terza modalità sono in grado di colonizzare nuovi territori e creare nuove popolazioni, mentre il polline ovviamente permette lo scambio di geni fra popolazioni già insediate. La riproduzione vegetativa è trascurabile in molte specie, ma in altre può costituire una modalità di riproduzione importantissima per la sopravvivenza della specie.
Da alcune misurazioni della velocità di colonizzazione di nuovi spazi desunta dalle datazioni con il radio-carbonio dei profili palinologici nei sedimenti lacustri, si è stimato che per le specie forestali più importanti come il faggio la velocità raggiunga i 200 metri all'anno. Questo implica una migrazione media dei semi di 8 km per ogni generazione di circa 40 anni.
Quest'elevatissa capacità di dispersione viene spiegata solo attraverso l'intervento di un agente dipersivo esterno, che nel caso delle fagacee potrebbe essere la ghiandaia Garrulus gladarius. Altri studi molto interessanti riguardano la simbiosi fra le specie con semi edibili e gli uccelli che ne assicurano la dispersione, o fenomeni di adattamento alla predazione sui semi come il ``masting''.
In prima approssimazione si può assumere che la distribuzione dei
propaguli nello spazio segua l'andamento di una curva normale
(gaussiana), che è sostanzialmente una esponenziale
negativa. L'86.5% dei propaguli si disperderebbe in tutte le
direzioni entro una distanza dal punto sorgente di 1
deviazione standard in un'area, chiamata area del vicinato
(neighbourhood area) pari a:
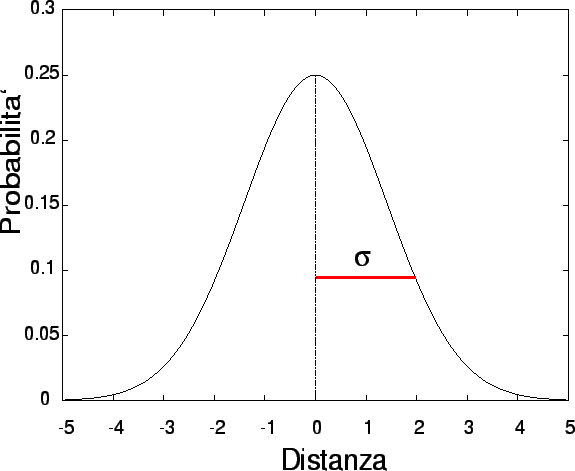
La varianza (![]() ) sarebbe la somma delle varianze delle
dispersione via polline, via seme e vegetativa
) sarebbe la somma delle varianze delle
dispersione via polline, via seme e vegetativa
dove t è il tasso di outcrossing, cioè 1 - s, dove s in questo caso è la proporzione di semi derivanti da autofecondazione.
La dispersione dei semi e l'insediamento delle piantine sono caratteristiche molto importanti per la fitness degli individui che dipendono da moltissimi fattori ecologici, biotici e abiotici.
In realtà si è visto da diversi dati raccolti sul campo che le assunzioni di dispersione del polline secondo una curva normale non sono valide. La dispersione del polline tende ad essere leptocurtica, cioè con una maggior frequenza di dati vicino alla media (al centro della distribuzione) rispetto a quanto previsto dalla curva normale.
Esistono numerosi modi per stimare il flusso genico (qui inteso come lo spostamento di geni fra ed entro le popolazioni) Distinguiamo in metodi:
Definizioni:
M = pianta madre;
D = pianta padre;
g = genotipo del figlio;
Assunzioni: Si assume che sia noto il genotipo di tutte le piante adulte con n potenziali padri, in una popolazione isolata senza immigrazione di polline esterno.
Obiettivi: Posto che un figlio della madre M abbia un genotipo multilocus (gi), stimare la probabilità condizionale che il padre putativo (j) sia il vero padre (Devlin et al, 1988).
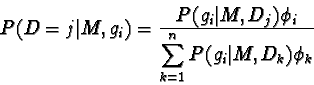
dove:
P( gi | M, Dj) sono le normali probabilità di segregazione date dalle leggi di Mendel, cioè le probabilità di ottenere un genotipo gi, dati i genotipi di M e di Dj.
e
![]() è la probabilità di paternità a priori, cioè la
probabilità che un figlio di M sia il frutto di una fecondazione
da parte del polline di Dj. Spesso viene considerata costante.
è la probabilità di paternità a priori, cioè la
probabilità che un figlio di M sia il frutto di una fecondazione
da parte del polline di Dj. Spesso viene considerata costante.
La probabilità di un padre putativo di essere il vero padre è data
da:
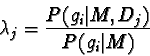
P( gi | M, Dj) sono di nuovo le normali probabilità di segregazione date dalle leggi di Mendel, cioè le probabilità di ottenere un genotipo gi, dati i genotipi di M e di Dj;
e
P(gi | M) sono le probabilità che un gamete della madre si incroci con un gamete preso a caso dalla popolazione per dare il genotipo gi. Dipende quindi dalle frequenze dell' allele pollinico nella popolazione. Intuitivamente si può capire che un potenziale padre ha più probabilità di essere il vero padre se l'allele dato al figlio è raro nella popolazione, mentre sarà meno probabile che sia il vero padre se l'allele dato al figlio è comune nella popolazione.
Di solito si confrontano le probabilità di tutti i possibili padri
calcolando il logaritmo di ![]() :
:
La paternità viene assegnata al padre che ha il LOD decisamente più alto degli altri. Se due potenziali padri hanno LOD simile non si attribuisce la paternità. Nel caso di marcatori non sufficientemente polimorfici, quest'ultima evenienza si presenta piuttosto spesso.
In questo caso la paternità viene frazionata fra tutti gli individui che presentano una probabilità ( P( gi | M, Dj)) maggiore di 0. La frazione di paternità assegnata al padre potenziale sarà proporzionale a P( gi | M, Dj) stessa.
Si è visto però che questo metodo, sebbene presenti dei vantaggi rispetto al metodo precedente, attribuisce artificialmente più paternità agli individui omozigoti rispetto a quelli eterozigoti.
È un metodo noto già da diverso tempo ma che praticamente si può usare solo quando si hanno a disposizione marcatori molto polimorfici, come i microsatelliti o SSR. Il vantaggio di questi marcatori sta nell'essere in grado di abbassare di molto la frazione di casi in cui la paternità rimane condivisa fra più potenziali padri, cioè P( gi | M, Dj) risulta uguale a 0 per molti individui. Vediamo in dettaglio questo metodo.